
Pharmacology is derived from the Greek words “Phamacon” which means drug and “logos” means knowledge. Pharmacology is a branch of science which deals with interaction of chemical molecules (drugs) within the living systems to affect, treat, or prevent diseases.

Rudolf Buchheim developed pharmacology as an experimental science and Oswald Schmiedeberg propounded some of the fundamental concepts in pharmacology. Pharmacology deals with the concept of Pharmacokinetics and Pharmacodynamics. Pharmacokinetics explains what the body does to the drug and pharmacodynamics explains what drug does to the body.
Pharmacokinetics (PK)
Pharmacokinetics is the study of drug movement in, through and out of the body. It consists of four phases. The entire process is called the ADME process of the body.
Adsorption: How will it get in?
Metabolism: How is it broken down?
Distribution: Where will it go?
Excretion: How does it leave?
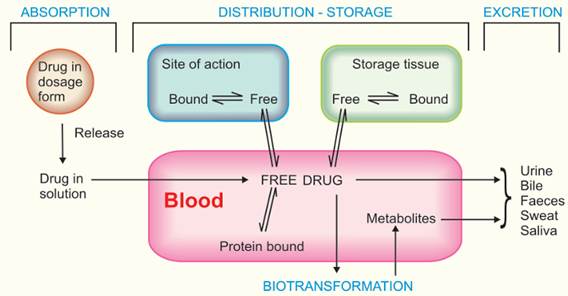
Source: Tripathi, K. D. (2013). Essentials of Medical Pharmacology (7th ed.). New Delhi: Jaypee Brothers Medical Publishers.
Absorption and Bioavailability
The first stage of pharmacokinetic is absorption. Absorption is the passage of drugs from the site of administration to the systemic circulation. Absorption depends upon:
- Aqueous solubility: The more the aqueous solubility, the faster the absorption is. For the poorly water-soluble drugs, the rate of absorption is governed by the dissolution rate.
- Concentration: A passive transport depends upon the concentration gradient, the more the concentrated solution the faster the absorption.
- Area of absorbing surface: If the larger the area the faster will be the absorption.
- Vascularity of absorbing surface: Increase in blood circulation helps in fast drug absorption. Blood flow helps to maintain the concentration gradient across the membrane.
- Route of administration: Each route has its own rate of drug absorption.
Intravenous (IV)>Inhalation >Sublingual/Buccal >Intramuscular (IM)>Subcutaneous (SC) >oral
↓ ↓ ↓ ↓ ↓ ↓
Highest/Intermediate V. Rapid Rapid Moderate to Rapid Slower than IM Slow
| Type of oral drugs | Site of absorption |
| Unionized lipid soluble | GIT |
| Acid drugs | Stomach |
| Basic drugs(ionized) | Duodenum |
Bioavailability refers to the rate and extent of absorption of a drug form dosage form. A drug is said to have 100% bioavailability when a fraction of drug reaches the systemic circulation in an unchanged form and becomes biologically available for therapeutic effect. It is possible after intravenous (IV) administration. Oral bioavailability is calculated by comparison of area under the plasma concentration-time curve (AUC) after IV dose of a drug with AUC at the same dose given by oral route.
(AUC) oral
Oral bioavailability = ————-X100
(AUC) IV
Oral bioavailability depends on the amount absorbed and amount metabolized before reaching the systemic circulation i.e. first pass metabolism.
Factors Affecting Bioavailability
- Molecular weight of drug: Drugs with lower molecular weight are absorbed easily through cell membrane. The more the absorption the greater will be the bioavailability and vice versa.
- Drug Formulation: The faster the dissolution the greater will be absorption and bioavailability. Liquid formulations have greater bioavailability than other formulations.
- Drug solubility: Drugs should be dissolved in body fluid before absorption. Higher the solubility, higher will be the bioavailability and vice versa.
- Chemical instability in gastric PH: Stability to the gastric PH increases the bioavailability and vice versa.
- First pass metabolism: Drug metabolized before reaching systemic circulation causes decrease in bioavailability. The degree of metabolism shows minimal or maximal decrease in bioavailability.
- Drug- food interaction: Food can alter the gastric emptying time, PH or interaction chemically. If the food enhances the dissolution or absorption, bioavailability will increase and if it hinders or delays absorption, bioavailability will decrease.
Distribution and Protein Binding
The second stage of pharmacokinetics is distribution. The distribution of the drugs is a dynamic process during which they pass from the central compartment in the tissue to reach the steady state plasma concentration to create the effect of the drug. Distribution depends upon mode of administration and the pK of the drug, its ability for binding to plasma protein, pH of the medium, tissue differences, blood flow and barriers. Drug distribution is initiated as free drug or unbound drug which is dissolved in plasma water or protein bound drug that exists as inactive. Distribution is a virtual space in which the drug is evenly distributed and is calculated in terms of volume of distribution (Vd), invasion (Penetration) and evaginate (release) rate constants. Drugs that get bound to plasma protein have low Vd and drugs that bound to extravascular tissues have high Vd.
Dose administered
Vd = ————————–
Plasma drug concentration
Most of the drugs possess physicochemical affinity for plasma protein. Plasma Protein Binding acts as a reservoir for sustained release of drugs causing prolonged action. The protein-drug complex cannot enter into capillaries and ends up into tissue due to large complex formation. Acidic drugs bind to plasma albumin and basic drugs bound to alpha-1 glycoprotein. The drug that is tightly bound has a longer duration of action and loosely bound or free drugs tend to act quickly and are excreted quickly. The significances of plasma protein binding are:
- Lower volume of distribution of highly Protein Binding drug due to restriction to vascular compartments.
- Higher degree of protein binding makes the drug long acting.
Side effects and adverse effects can occur when drugs bind to other sites than the targeted site.
Metabolism and the Cytochrome P450 System
After the distribution phase of pharmacokinetics, drugs are broken down by a process known as metabolism. Metabolism is also known as biotransformation or detoxification. Drugs undergo various chemical changes to make a compound which is easily excreted from the body. For example: lipid soluble and unionized compounds into water- soluble and ionized compounds to prevent reabsorption. The primary site for drug metabolism is liver and others are kidney, intestine, lungs and plasma. Metabolism of drugs leads to:
- Inactivation of drug: Most drugs and their active metabolites are converted to less active or inactive metabolites. For example phenobarbital, propranolol, etc.
- Active metabolites from an active drug: Many drugs are converted to one or more active metabolites. For example diazepam, amitriptyline, etc.
- Activation of inactive drug: Few drugs are in inactive form which need conversion in the body to one or more active metabolites. For example levodopa, enalapril, etc.
There is two mechanism of biotransformation:
- Phase I (Non-synthetic or Functionalization)
In this phase the inactive drug is changed into an active form of drug (pro-drug). It involves oxidation, reduction, hydrolysis reaction to slightly increase the hydrophilicity. Metabolites may be active or inactive.
- Phase II (Synthetic or Conjugation)
The phase II metabolism involves reactions that couple the drug molecules with other molecules in a conjugation process, which cause drug compounds pharmacologically inert and water- soluble so that it can be easily excreted. It involves glucuronide, sulfate, N-acetylation, methylation and conjugation with glutathione, and conjugation with amino acid to largely increase hydrophilicity.
Hepatic smooth endoplasmic reticulum contains cytochrome P450 enzyme, is a primary liver enzyme which metabolizes drugs. Cytochrome P450 are named so because they are bound to membranes within a cell (cyto) and contain a heme pigment (chrome and P) which absorbs light at a wavelength of 450nm when exposed to carbon-monoxide. Cytochrome P450 enzymes are the important enzymes for metabolism of many drugs. More than 50 enzymes are present in this group and six of them (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5) metabolize 90% of the drug. CYP3A4 and CYP2D6 are the most significant enzymes. Cytochrome P450 enzymes are essential for the production of cholesterol, steroids, prostacyclin’s and thromboxane A2. These cytochrome enzymes are mostly expressed in the liver, but also occur in the small intestine (reducing drug bioavailability), lungs, placenta and kidneys. Phase I metabolism is mainly carried out by this enzyme. This enzyme either induces the metabolism or inhibits it.
- Enzyme Induction: In this process one drug induces the activity of metabolizing enzymes, leading to increased metabolism of another drug. It causes a decrease in therapeutic activity of drugs and increases drug toxicity. For example: If Drug A induces the CYP450 enzyme and Drug B is taken together then Drug B will be metabolized faster and the therapeutic effect of Drug B will be reduced.
- Enzyme inhibition: It refers to decrease in enzyme activity and leads to reduced metabolism of drugs. This can cause serious adverse effects. For example: If Drug A inhibits the enzyme then drug B which is taken together will be metabolized slowly, increasing the plasma concentration of Drug B causing toxicity.
Excretion and Drug Half-Life
Excretion is the last pharmacokinetic stage of a drug. It is also known as elimination. The two principal organs responsible for drug elimination are the kidney and the liver. The kidney is the primary site for removal of a drug in a chemically unaltered or unchanged form as well as for metabolites. The lungs are responsible for the elimination of high vapor pressure (alcohol, gaseous anesthetics, etc.). Other ways of eliminating are bile & feces, saliva & sweat, milk and skin. The kidney is the main organ for excretion. It consists of glomerular filtration, passive tubular reabsorption and active tubular secretion. The excretion of the drug is determined by calculating the clearance (Cl) rate. The clearance of a drug is the theoretical volume of plasma from which the drug is removed completely in unit time.
Calculated as Cl= Rate of elimination/ Plasma drug concentration
Half- Life refers to the rate at which 50 % of a drug is eliminated from the body, it affects the duration of the therapeutic effect of a medication. The ADME processes influence a drug’s half- life. The distribution (Vd) is directly proportional to half-life and excretion (clearance) is inversely proportional to half-life.
I.e. Half-life (t1/2) = (0.693 x Vd) / Cl
Half-life impacts the time taken by a drug to reach steady-state level. Some drugs have short half- life and some have significantly larger. Any impairment in either metabolizing organ (liver) or elimination organ (kidney) can significantly alter medication dosing, frequency of doses, therapeutic effects and half-life of the drug.
The importances of half- life study are:
- Helps to predict the time taken by drug to leave the body completely after discontinuation.
- Helps to determine duration of drug action. If it is a short acting or long acting drug.
- Helps in frequency of dosing for repeatedly given drugs.
- Helps in determining the loading dose.
- Helps to prevent drug accumulation and toxicity.
Pharmacodynamics (PD)
Pharmacodynamics is the branch of pharmacology concerned with the effects of drugs and mechanism of their action. It explains the type of response produced by drugs and how and where the effect is produced in the body. When the drug or chemical substance enters the body system, multiple changes in and interference with the functioning of the cell occurs. There are two basic concepts that describe the pharmacodynamics action of all drugs; affinity and efficacy. Affinity means molecular forces that keep the drug associated with the target long enough for a biological effect to occur and efficacy is the intrinsic property of the drug that causes the target to change in response to drug binding. It shows strong drug-receptor interaction.
Drug-Receptor Interactions (The Lock and Key Model)
Receptor sites are the specific site on a cell membrane where a drug acts. The interaction between the chemical and the receptor site affects enzyme systems within the cell. To further understand, the lock and key model is important. The lock is the specific receptor site and key is a specific chemical substance which has a perfect fit for the lock. Emil Fischer proposed the Lock and Key model of enzyme action. It was proposed for the fact that enzymes are very specific for the substrate. This model hypothesized that active sites of enzymes have the same conformation as substrate which allows binding of substrate in the active site of the enzyme just like key fits in the lock. Product is generated after the enzyme acts on the substrate.
Limitation of lock and key model
- There is change in conformation of the active site of the enzyme upon binding of substrate.
- It doesn’t explain the condition for multiple substrates binding to the enzyme.
- Enzyme-substrate complex stabilization in transition state isn’t explained.
To explain all the limitations the induced fit model was later introduced.
There are three types of drug actions on receptors:
Agonist: An agonist is a substance that binds to a receptor and causes a desired effect. It has both affinity and intrinsic activity. For example, in asthmatic patients Salbutamol binds to beta-2 receptors in bronchial smooth muscle causing bronchodilation.
Antagonists: An antagonist is the substance that binds to a receptor and blocks the action of agonist effect at the binding site. It has affinity but no intrinsic activity. For example, naloxone binds to opioid receptors and deactivates the receptor preventing opioid overdose. Antagonists are of two types:
- Competitive Antagonism: There are two types of competitive antagonism: Competitive reversible antagonism and Competitive irreversible antagonism. Competitive reversible antagonism occurs when a drug (antagonist) binds to the same receptor site as an agonist. The binding is weak and reversible. If the concentration of agonist is increased there will be a shift in the dose-response curve to the right without reducing the maximum response. Competitive irreversible antagonism binds covalently to the active site of a receptor and permanently inactivates it. Effects cannot be overcome even by increasing the concentration of the agonist.
- Non- competitive Antagonism: This antagonism occurs when the antagonist binds irreversibly or allosterically to a different site than agonist’s binding site.
Inverse Agonists: A drug that binds to a receptor but produces an opposite effect than agonists. For example, antihistamines act as inverse agonists at H1 receptors, reducing receptor activity and decreasing allergic symptoms.
Therapeutic Index and Drug Safety
Definition: The therapeutic index of a drug is the ratio of the dose that exerts toxicity (TD50) or lethality (LD50) in 50% of the population to the dose that exerts a therapeutic or effective response (ED50) in 50% of the population.
Median Lethal Dose (LD50) OR Median Toxic Dose (TD50)
Therapeutic Index = ———————————————————————-
Median Effective Dose (ED50)
Where Median effective dose (ED50) is the dose which produces the specified effect in 50% individuals and median lethal dose (LD50) or median toxic dose (TD50) is the dose which kills or shows toxic effect to 50% of the recipients. These are measured in animal studies that represent the safety margin of a particular drug in clinical use. The higher the therapeutic index value, the wider the margin between effective and toxic doses.
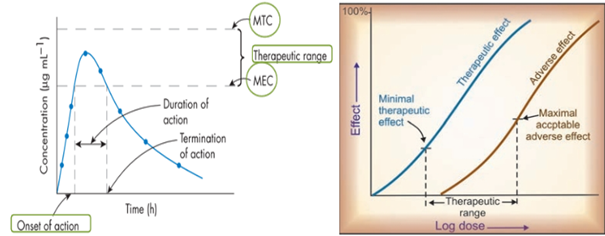
Source: Tripathi, K. D. (2013). Essentials of Medical Pharmacology (7th ed.). New Delhi: Jaypee Brothers Medical Publishers.
The drug is said to be safe when its plasma drug concentration- time profile is below the Maximum Therapeutic Concentration (MTC) and said to be effective if the curve is above the Minimum Effective Concentration (MEC).
Conclusion
Pharmacokinetics and pharmacodynamics are closely related, pharmacokinetics determines the concentration of drug reaching the site of action while pharmacodynamics determines the pharmacological effect produced by that concentration of drug. Pharmacokinetic processes mostly occur in the liver, people with liver disease are often a contradiction or a reason to cause caution during drug administration. Dose optimization should be done for the patient with complications. Additionally, understanding these principle concepts of pharmacology allows optimizing drug therapy, minimizing adverse effects, and ensuring patient safety and rational and effective use of medications in clinical practice.
References
- Absorption of Drugs – Biopharmaceutics and Pharmacokinetics. (n.d.). Pharmacy180.com. https://www.pharmacy180.com/article/absorption-of-drugs-2448/
- Ernstmeyer, K., & Christman, E. (2023). Pharmacokinetics & pharmacodynamics. National Library of Medicine; Chippewa Valley Technical College. https://www.ncbi.nlm.nih.gov/books/NBK595006/
- Lynch, T., & Price, A. (2007). The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. American Family Physician, 76(3), 391–396.
- Pharm, B., Ahmedabad, Bengaluru, Bhopal, Bhubaneswar, Chennai, Dehradun, Ernakulam, Jaipur, H., Jalandhar, Kolkata, Nagpur, Patna, Pune, & Rohtak. (n.d.). BIOPHARMACEUTICS & PHARMACOKINETICS. Retrieved March 11, 2026, from https://depthofbiology.com/wp-content/uploads/2024/08/PharmaLite.in-Biopharmaceutics-Pharmacokinetics-Thakur-1.pdf
- Tripathi, K. D. (2013). Essentials of medical pharmacology (pp. 10–60). Jaypee Brothers Medical Publishers (P) Ltd.