
Molecular docking is an important method for structural molecular biology and computer-aided drug development. Molecular docking often occurs between a small molecule and a target macromolecule. This is commonly known as ligand-protein docking, although there is growing interest in protein-protein docking.
The docking methodology predicts the experimental binding modes and affinities of small molecules within the binding site of specific receptor targets. It is currently used as a standard computational tool in drug design for lead compound optimization and virtual screening studies to find novel biologically active molecules. A search algorithm and an energy-scoring function are the primary tools used in docking methods to generate and evaluate ligand poses.
The automated molecular docking program aims to anticipate molecular recognition through both structural and energetic analysis. Docking small molecules to biological targets involves exploring several ligand poses in the target’s groove or pocket to determine the best binding shape.
Molecular docking
Molecular docking is a computational tool for enhancing molecular interactions and predicting binding affinity. The molecular docking process simulates conformation according to complementarity and pre-organization to predict and obtain the binding affinity and interaction mode of ligand and target. In general, two molecular models are used for molecular docking. They are:
Lock and key model:
In 1890, Emil Fischer created the “lock-and-key model” to explain biological processes. The substrate is introduced into the active site of a macromolecule, similar to how a key is inserted in a lock. Biological locks require precise stereochemical properties to function properly. It refers to the rigid docking process in which ligands find the correct orientation to fit in the complementary shape of the target protein.
Induced fit model:
Daniel Kochland presented the “Induced fit theory” in 1958. The fundamental idea is that the ligand and the target adjust to one another during character recognition by making small conformational changes until an ideal match is found. It involves the flexible docking process in which ligand and protein adjust their conformation to fit together.

Types of molecular docking
Rigid docking:
In rigid docking, both the receptor and ligand molecules remain stationary. Both the ligand and the protein are rigid entities, and only the three translational and three rotational degrees of freedom are taken into account during sampling. This approach is similar to the “lock-key” binding model and is predominantly used for protein-protein docking when the number of conformational degrees of freedom is too large to sample. Molecules can only change their position during rigid docking, not their spatial shape. Docking is carried out. Based on a scoring function, we aim to convert one of the molecules to a 3D environment that best matches the other molecules. The ligand’s conformation can occur without or with receptor binding activity.
Rigid body docking generates several conformations with favorable surface complementarity, which are then reranked based on the free energy of approximation. Rigid-body docking simulation is a quick way to screen a small molecule database for virtual screening. When compared to crystallographic structures, it has a comparatively high accuracy. This accuracy would increase even more if we performed a study of the top outcomes from rigid-body docking simulations, using an empirical scoring model.
Flexible docking
Flexible docking approaches allow for several ligand or receptor configurations, as well as both molecules at once, but require more computing effort. Standard virtual docking experiments include freely docking ligands onto stiff receptors. Side-chain flexibility is critical for ligand-protein interactions, as evidenced by recent research. The receptor can vary its binding location based on the ligand’s orientation. The ligand moves in a 6 + N-dimensional space of translation, rotation, and conformation in the receptor’s anisotropic environment.
So, flexible docking allows for the movement of both the ligand and the receptor. It is conformationally flexible. Energy is calculated during each spin. The surface cell occupancy for each configuration is computed. Finally, the optimal binding pose is chosen.
Search methods of Molecular docking
There are mainly two types of search methods. They include:
Systematic method:
The search results are deterministic, but the quality of the solution is determined by the granularity of the search space sampling. It is further divided into three subtypes: The three subtypes of systematic approaches are as follows:
- Conformational search: The torsional (dihedral), translational, and rotational degrees of freedom of the ligand’s structural parameter are gradually altered.
- Fragmentation: During the molecular docking process, numerous fragments may be docked together to create bonds, or the fragments may be anchored individually, with the first fragment docked initially and the following fragments constructed outward in steps from the initial bound point.
- Database Search: Using this technique, it is possible to generate several plausible conformations of every small molecule previously recorded in the database and dock them as hard bodies.
Stochastic approaches
Stochastic approaches depend on an element of randomness, therefore the outcome changes. Different types of this approaches include:
- Monte Carlo: This method includes randomly inserting ligands in the receptor binding site, scoring them, and then creating a new configuration.
- Genetic algorithm: It begins with a population of postures, where the “gene” describes the configuration and placement of the receptor, and the score is the “fitness.” Perform fittest transformations, hybrids, and so on to create the next generation and then repeat the agreement.
- Tabu search: It works by striking limits that facilitate the investigation of a new configuration by limiting the previously exposed parts of the ligands’ conformational space from being studied again.
Search algorithms for Molecular docking
Different types of search algorithms are used in the molecular docking process. Some are as follows
Fast shape matching (SM)
Shape-matching algorithms consider the geometric overlap between two molecules.
Several algorithms are used to align ligands and receptors. This strategy involves searching the macromolecular surface for potential protein binding sites. Furthermore, SM-specific algorithms identify probable conformations of expected binding sites.
Incremental construction (IC)
This approach separates the ligand into fragments that bind independently in the receptor location. After docking the fragments, they are fused. This fragmentation enables the program to consider ligand flexibility. Docked rigid fragments act as “anchors” and are connected by flexible ligands with rotatable links. In this technique, the ligand is gradually “constructed” within the receptor’s binding region.
Monte Carlo simulations (MC)
Monte Carlo implementations take the ligand as a whole and make random changes to its translation, rotation, and torsion angles. After each move, the structure is minimized and the energy of the new structure is calculated. Minimizing before considering the Metropolis criterion was found to improve convergence.
In this approach, the ligand is typically put at random in the binding site. To enhance the likelihood of obtaining the global energy minimum, the simulation may involve multiple cycles. The first cycle occurs at high temperatures, followed by successively lower temperatures (simulated annealing MC). Usually, each cycle begins with the lowest energy from the previous cycle.
Simulated annealing (SA)
Simulated annealing simulates biomolecular systems using dynamic simulations. Each docking conformation undergoes a simulation that gradually decreases temperature at regular intervals throughout each cycle. Compared to MC, this method may provide more accurate results by taking into account the conformational state and flexibility of both proteins and ligands in various thermodynamic states across time.
Distance Geometry
This search strategy utilizes information expressed as intra- and intermolecular distances. Assembling these distances allows for the determination of structures and conformations.
Tabu search (TS)
Tabu search (TS) is an iterative process used to solve optimization problems. Glover invented and detailed this method, which has successfully solved numerous challenging optimization issues. The Tabu search docking approach is highly accurate, preventing simulations from becoming locked in local minima and avoiding known minimal energy conformations.
Scoring functions for Molecular docking
To rank ligand orientations/conformations, a scoring method evaluates the binding strength of each potential complex. An ideal scoring function would give the empirically determined binding mode the highest score. By determining a ligand’s binding mode, scientists can obtain a deeper knowledge of the molecular mechanism and build efficient drugs by changing the protein or ligand. A scoring function can also predict the absolute binding affinity between a protein and ligand, similar to the first application. This is very significant in lead optimization.
Different scoring functions include:
Force field scoring function
Force field-based functions are composed of a sum of energy terms from a classical force field, typically taking into account the interaction energies of the protein-ligand complex (non-bonded terms) and the internal ligand energy (bonded and non-bonded terms). It is based on physical atomic interactions, such as van der Waals (VDW), electrostatic interactions, and bond stretching/bending/torsional forces. According to fundamental physics, force field functions and parameters are often obtained using experimental data and ab initio quantum mechanical computations. The treatment of solvents in ligand binding poses a significant barrier for force field scoring systems, despite their apparent physical meaning.
Empirical scoring function
It is based on repetitive linear relapse analysis of a preset set of complex structures containing protein-ligand complexes with known binding affinities, functional groups, and some form of interaction. Examples include the N-O hydrogen link, the O-O hydrogen bond, salt scaffolding, aromatic ring stacking, and so on. Empirical scoring functions are generated to replicate experimental affinity data, with the idea that the free energy of binding can be correlated to a set of unrelated variables. Van der Waals, electrostatic, hydrogen bond, desolvation, entropy, hydrophobicity, and other empirical energy terms are added togetherEmpirical scoring functions are faster at calculating binding scores than force field scoring functions due to their simpler energy factors.
Knowledge-based scoring function
These methods assume that statistically more explored ligand-protein contacts are associated with favorable interactions. When evaluating a pose, all ligand-protein atom pairs are added up, yielding the pose’s score. Knowledge-based scoring functions are sometimes known as statistical-potential scoring functions. It uses energy potentials based on experimentally known atomic structures. The idea underlying knowledge-based scoring functions is straightforward: Pairwise potentials are derived from the frequency of atom pairs in a database using the inverse Boltzmann relation.
Consensus scoring
There are numerous scoring functions available, but none are perfect in terms of accuracy and broad applicability. Each scoring function has both advantages and disadvantages. The consensus scoring technique combines scores from numerous scoring functions to improve the likelihood of the right solutions, balancing the benefits and drawbacks of each. Designing an acceptable consensus scoring system for individual scores is crucial for identifying true modes/binders.
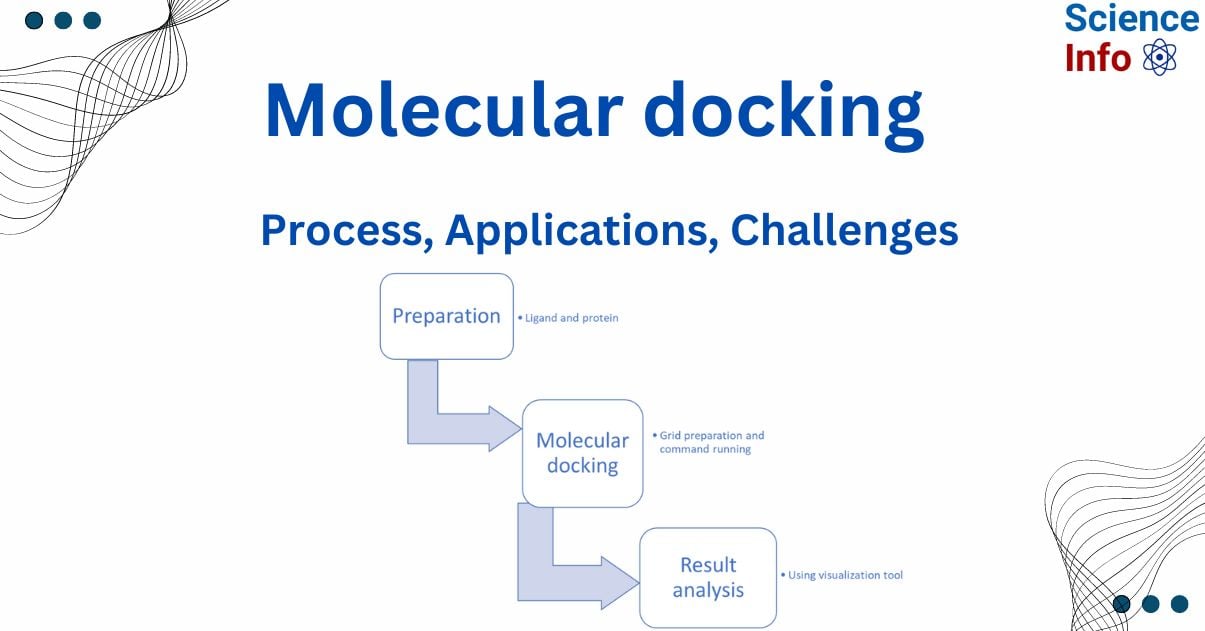
Process of molecular docking

Retrieval and preparation of target molecule
The features of the chosen protein structure impact docking outcomes. Advancements in structure determination technologies, such as X-ray crystallography, NMR, and cryo-EM, have led to an increase in the number of proteins with known three-dimensional (3D) structures, which are now available in public databases such as the Protein Data Bank (PDB). The first step in docking is retrieving the 3D structure of the protein, preferably with a ligand, from the PDB. It is recommended to use 3D structures with high resolution (˂ 2Å) or those bound by high-affinity ligands.
The features of the chosen protein structure impact docking outcomes. Advancements in structure determination technologies, such as X-ray crystallography, NMR, and cryo-EM, have led to an increase in the number of proteins with known three-dimensional (3D) structures, which are now available in public databases such as the Protein Data Bank (PDB). The first step in docking is retrieving the 3D structure of the protein, preferably with a ligand, from the PDB. It is recommended to use 3D structures with high resolution (˂ 2Å) or those bound by high-affinity ligands.
Preparation of ligand
Ligand structures are generated using programs like ChemDraw or retrieved from databases such as PubChem and ZINC. To use these structures in docking, energy minimization should be performed.
Docking
After preparing the target and ligand, the binding site should be identified and constrained. This step can be completed manually or using the coordinates of a ligand bound to the protein. The software can determine the most likely binding site. The grid maps the binding area, which serves as the center for docking calculations. The grid is a box with known dimensions divided into small squares, where probe atoms represent probable interactions. The resolution and size of the grid impact docking results.
Then docking can be performed by using certain commands depending on the software.
Analysis of docking result
Docking results from autodock can be analyzed by using various visualization tools like BioVia Discovery Studio.
Validation of docking
Validation of the docking procedure is necessary, as with any other technique. Docking data are confirmed by redocking reference ligands with targets and comparing RMSD values, binding posture, binding affinity, and predicted binding coverage to earlier results.
Applications of molecular docking
- Modeling the structure of a protein-ligand complex helps understand the interaction between a potential curative chemical (the ligand) and its target protein.
- Computer-aided docking may explore the motion space of protein-ligand complexes, resulting in a stable configuration that accurately represents the complex’s structure.
- Protein-ligand docking can predict which chemicals are degradable by enzymes. It can help determine a preferred place.
- Reverse docking predicts the biological target of a given chemical. This approach is effective for identifying and profiling computational targets.
- Virtual screening identifies hits and lead compounds in molecular databases using scoring systems.
- Docking can also be used to predict small molecule attributes such as absorption, distribution, metabolism, excretion, and toxicity (ADMET). The projected ADMET attributes can be utilized to rule out compounds with undesirable features early in the drug discovery process.
- Molecular docking is an important tool for predicting drug binding characteristics to nucleic acids. This material demonstrates the link between a drug’s molecular structure and its cytotoxicity. Keeping this in mind, medicinal chemists are continually working to understand the underlying anticancer mechanism of medications at the molecular level by studying the interaction pattern of nucleic acids and pharmaceuticals.
- Molecular docking can be used with molecular dynamic simulations to investigate the dynamic behavior of protein-ligand complexes. The simulations can assist explain the conformational changes that occur when ligands bind to the complex, as well as its stability.
- Molecular docking can also be used to determine the structure of unknown proteins. Docking can be used to anticipate the binding mechanisms of small molecules to proteins and then construct a protein homology model based on those predictions.
Challenges during molecular docking
- During docking the handling of flexible proteins presents a significant problem. A biomolecule/protein changes conformation based on the ligand to which it binds. This indicates that docking stiff receptors results in a single receptor conformation. However, when docking is performed using a flexible receptor, the ligands may require many receptor conformations to interact. The most ignored component of molecular docking studies is the many conformational states of proteins.
- The ligand preparation has a significant impact on the docking outcomes since ligand recognition by any biomolecule is dependent on 3-dimensional orientation and electrostatic interactions. This confirms the importance of ligand structure and ligand preparation.
- Another problem in docking is the inaccuracy of the scoring function. Just as search algorithms can provide the best conformation, scoring functions should be able to distinguish real binding modes from all other parallel forms. A hypothetical scoring algorithm would be computationally efficient, but unsuitable for analyzing many binding modes. When there is accuracy, scoring functions generate several options for evaluating ligand affinity. Scoring algorithms overlook physical phenomena like as entropy and electrostatic interactions. As a result, the fundamental constraint in molecular docking programming is a lack of an appropriate scoring function that is both accurate and fast.
References
- https://www.omicsonline.org/open-access/molecular-docking-approaches-types-applications-and-basic-challenges-2155-9872-1000356.php?aid=88070#14
- https://pubs.rsc.org/en/content/articlelanding/2010/cp/c0cp00151a/unauth
- https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.10115
- https://link.springer.com/article/10.1007/s12539-019-00327-w
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4783878/
- https://link.springer.com/article/10.1007/s40484-019-0172-y
- https://www.intechopen.com/chapters/70982
- https://jabonline.in/admin/php/uploads/443_pdf.pdf
- https://sites.ualberta.ca/~pwinter/Molecular_Docking_Tutorial.pdf